



| ELISA kit |
| Clinical diagnosis |
| Trfia |
| ANTIBODIES |
| CLIA kits |
| Proteins |
| New Products |
| > Human |
| > Canine |
| > Bovine |
| > Equine |
| > Fish |
| > Mouse |
| > Rat |
| > Chicken |
| > Porcine |
| > Rabbit |
| > Goat |
| > Simian |
| > General |
| > Cavia |
| > Caprine |
| > Sheep |
| > Cattle |

Recombinant Human α-Galactosidase/GLA
Instruction Manual!
| Product name: | Recombinant Human α-Galactosidase/GLA |
| Source: | Human Cells |
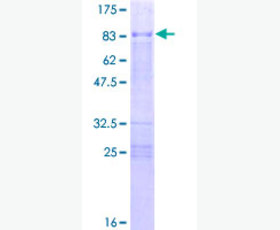
| Purity: | Greater than 95% as determined by reducing SDS-PAGE. |
| Buffer Formulation: | Supplied as a 0.2 μm filtered solution of 20mM TrisHCl, 150mM NaCl, pH 8.0. |
| Applications: | Applications:SDS-PAGE; WB; ELISA; IP. |
| Storage: | Avoid repeated freeze/thaw cycles. Store at 2-8 oC for one month. Aliquot and store at -80 oC for 12 months. |
| UOM: | 100ug/50ug/200ug/1mg/1g |
| Source | Human Cells |
| Description | Recombinant Human alpha-Galactosidase is produced by our Mammalian expression system and the target gene encoding Leu32-Leu429 is expressed with a 6His tag at the C-terminus. |
| Names | Alpha-Galactosidase A, Alpha-D-Galactosidase A, Alpha-D-Galactoside Galactohydrolase, Melibiase, Agalsidase, GLA |
| Accession # | P06280 |
| Formulation | Supplied as a 0.2 μm filtered solution of 20mM TrisHCl, 150mM NaCl, pH 8.0. |
| Shipping |
The product is shipped on dry ice/ice packs. |
| Storage |
Store at < -20°C, stable for 6 months after receipt. Please minimize freeze-thaw cycles. |
| Purity |
Greater than 95% as determined by reducing SDS-PAGE. |
| Endotoxin | Less than 0.1 ng/µg (1 IEU/µg) as determined by LAL test. |
| Amino Acid Sequence |
LDNGLARTPTMGWLHWERFMCNLDCQEEPDSCISEKLFMEMAELMVSEGWKDAGYEYLCIDDCWM APQRDSEGRLQADPQRFPHGIRQLANYVHSKGLKLGIYADVGNKTCAGFPGSFGYYDIDAQTFAD WGVDLLKFDGCYCDSLENLADGYKHMSLALNRTGRSIVYSCEWPLYMWPFQKPNYTEIRQYCNHW RNFADIDDSWKSIKSILDWTSFNQERIVDVAGPGGWNDPDMLVIGNFGLSWNQQVTQMALWAIMA APLFMSNDLRHISPQAKALLQDKDVIAINQDPLGKQGYQLRQGDNFEVWERPLSGLAWAVAMINR QEIGGPRSYTIAVASLGKGVACNPACFITQLLPVKRKLGFYEWTSRLRSHINPTGTVLLQLENTM QMSLKDLLVDHHHHHH
|
| Background | α-Galactosidase A is a homodimeric glycoprotein that belongs to the glycosyl hydrolase 27 family. It is a lysosomal enzyme and used as a long-term enzyme replacement therapy in patients with a confirmed diagnosis of Fabry disease. α-Galactosidase A can hydrolyze terminal α-galactosyl moieties from glycolipids and glycoproteins and catalyze the hydrolysis of melibiose into galactose and glucose. Defects α-Galactosidase A are the cause of Fabry disease (FD) which is a rare X-linked sphingolipidosis disease with glycolipid accumulates in many tissues. The disease consists of an inborn error of glycosphingolipid catabolism. FD patients show systemic accumulation of globotriaoslyceramide (Gb3) and related glycosphingolipids in the plasma and cellular lysosomes throughout the body. Patients may show ocular deposits, febrile episodes, and burning pain in the extremities. Death results from renal failure, cardiac or cerebral complications of hypertension or other vascular disease. |








