



| ELISA kit |
| Clinical diagnosis |
| Trfia |
| ANTIBODIES |
| CLIA kits |
| Proteins |
| New Products |
| > Human |
| > Canine |
| > Bovine |
| > Equine |
| > Fish |
| > Mouse |
| > Rat |
| > Chicken |
| > Porcine |
| > Rabbit |
| > Goat |
| > Simian |
| > General |
| > Cavia |
| > Caprine |
| > Sheep |
| > Cattle |

Recombinant Human β-Ureidopropionase/UPB1
Instruction Manual!
| Product name: | Recombinant Human β-Ureidopropionase/UPB1 |
| Source: | E.coli |
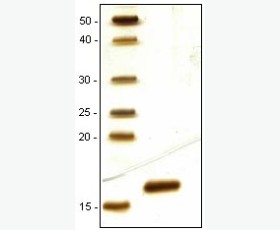
| Purity: | Greater than 95% as determined by reducing SDS-PAGE. |
| Buffer Formulation: | Supplied as a 0.2 μm filtered solution of 20mM PB, 150mM NaCl, pH 7.4. |
| Applications: | Applications:SDS-PAGE; WB; ELISA; IP. |
| Storage: | Avoid repeated freeze/thaw cycles. Store at 2-8 oC for one month. Aliquot and store at -80 oC for 12 months. |
| UOM: | 100ug/50ug/200ug/1mg/1g |
| Source | E.coli |
| Description | Recombinant Human beta-Ureidopropionase is produced by our E.coli expression system and the target gene encoding Met1-Glu384 is expressed with a 6His tag at the C-terminus. |
| Names | Beta-Ureidopropionase, BUP-1, Beta-Alanine Synthase, N-Carbamoyl-Beta-Alanine Amidohydrolase, UPB1, BUP1 |
| Accession # | Q9UBR1 |
| Formulation | Supplied as a 0.2 μm filtered solution of 20mM PB, 150mM NaCl, pH 7.4. |
| Shipping |
The product is shipped on dry ice/ice packs. |
| Storage |
Store at < -20°C, stable for 6 months after receipt. Please minimize freeze-thaw cycles. |
| Purity |
Greater than 95% as determined by reducing SDS-PAGE. |
| Endotoxin | Less than 0.1 ng/µg (1 IEU/µg) as determined by LAL test. |
| Amino Acid Sequence |
MAGAEWKSLEECLEKHLPLPDLQEVKRVLYGKELRKLDLPREAFEAASREDFELQGYAFEAAEEQ LRRPRIVHVGLVQNRIPLPANAPVAEQVSALHRRIKAIVEVAAMCGVNIICFQEAWTMPFAFCTR EKLPWTEFAESAEDGPTTRFCQKLAKNHDMVVVSPILERDSEHGDVLWNTAVVISNSGAVLGKTR KNHIPRVGDFNESTYYMEGNLGHPVFQTQFGRIAVNICYGRHHPLNWLMYSINGAEIIFNPSATI GALSESLWPIEARNAAIANHCFTCAINRVGTEHFPNEFTSGDGKKAHQDFGYFYGSSYVAAPDSS RTPGLSRSRDGLLVAKLDLNLCQQVNDVWNFKMTGRYEMYARELAEAVKSNYSPTIVKEVEHHHH HH
|
| Background | β-Ureidopropionase is a cytoplasmic protein which belongs to the CN hydrolase family of BUP subfamily. β-Ureidopropionase binds one zinc ion per subunit, catalyzes the last step in the pyrimidine degradation pathway. β-Ureidopropionase can convert N-carbamyl-beta-aminoisobutyric acid and N-carbamyl-beta-alanine to beta-aminoisobutyric acid and beta-alanine, ammonia and carbon dioxide, respectively. The pyrimidine bases uracil and thymine are degraded via the consecutive action of dihydropyrimidine dehydrogenase (DHPDH), dihydropyrimidinase (DHP) and beta-ureidopropionase (UP) to beta-alanine and beta aminoisobutyric acid, respectively. Defects in β-Ureidopropionase are the cause of β-Ureidopropionase deficiency that is characterized by muscular hypotonia, dystonic movements, scoliosis, microcephaly and severe developmental delay. |








